New publication for NCMM researchers: novel insights into cell death-inducing signals activated by ER stress
The new study, published this week in the open access journal Cell Communication and Signaling, was led by the autophagy team at NCMM.


The researchers’ findings have important implications for cancer-related therapies that work by inducing a process in cells known as the ‘unfolded protein response’. First author of the paper, Paula Lindner, is a joint PhD student at NCMM and our Nordic EMBL Partnership sister node DANDRITE (Aarhus, Denmark), and the publication comes as a product of collaborative work between NCMM, DANDRITE, the University of Copenhagen and Aarhus University.
Keeping proteins folded
Each one of our cells contains thousands of protein molecules that work together to control various aspects of cell behavior and function, including cell division, movement, and even the decision of a cell to survive or die. To perform their jobs properly, it is vital that each of these protein molecules is correctly and precisely folded. Failure to regulate protein folding can lead to dysfunctional proteins and may ultimately be toxic to the cell.
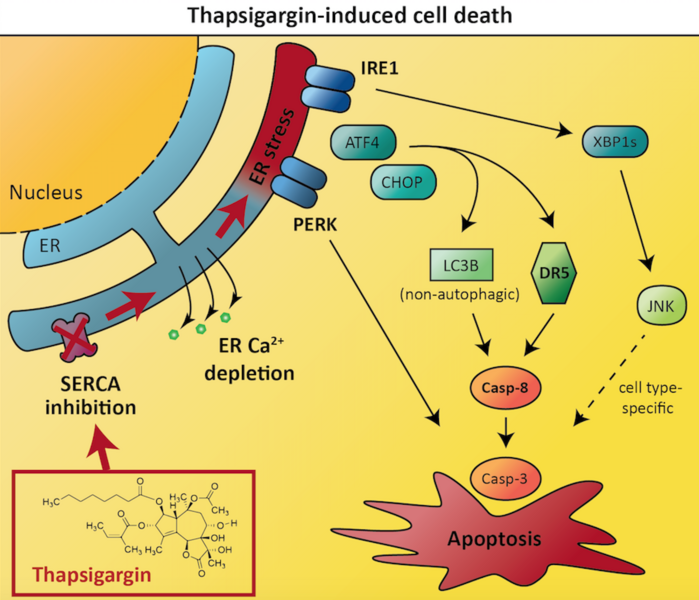
New proteins are constantly being synthesized by our cells – just as old proteins are constantly degraded to make way for new ones. The endoplasmic reticulum (ER) is a membrane-enclosed network of tubules within our cells that works to ensure all newly synthesized proteins are properly folded. By performing this task, the ER performs an essential house-keeping task and makes sure that unfolded, faulty proteins do not accumulate in the cell.
However, in some instances, the number of new proteins produced by the cell might exceed the ER’s capacity to oversee correct protein folding. Under these circumstances, a process known as ‘ER stress’ occurs. In an attempt to keep misfolded protein levels to a minimum, the cell reacts to ER stress by triggering a type of internal emergency alarm known as the unfolded protein response (UPR).
In the first instance, the UPR involves trying to re-establish the balance of normal vs misfolded proteins: synthesis of new proteins is shut down to relieve the over-worked ER, and the pathways that induce protein degradation are amplified to remove misfolded proteins. However, if this first stage of the UPR fails to restore healthy conditions within the cell then stage 2 is activated, and the ‘abandon ship’ signal is triggered. The cell activates a type of programmed cell death known as apoptosis, which leads to the tightly controlled destruction of the cell in a way that ensures nearby cells are not exposed to toxic cell debris.
Activating the UPR in cancer cells
This system of triggering apoptosis via ER stress and the UPR represents one of the ways scientists are trying to treat diseases such as cancer. By exposing cancer cells to drugs that induce ER stress, it is hoped that the UPR will induce cancer cell death and, therefore, the shrinking of tumors.
However, current understanding of the signaling mechanisms involved in UPR-mediated activation of cell death is incomplete, which restricts researchers’ ability to design drugs that can effectively activate these processes in cancer cells.
In their recent study, the autophagy team at NCMM aimed to address this problem and improve our understanding of ER stress and the UPR in cancer cells. They investigated an ER stressor drug called thapsigargin. This compound is known to induce UPR-mediated cell death, but a detailed understanding of the mechanisms involved is lacking.
The researchers aimed to identify which cellular proteins were required for thapsigargin to induce cancer cell death. To achieve this, they exposed human prostate and colon cancer cell lines to thapsigargin, and combined this treatment with inhibition or inactivation of a range of different proteins within the cancer cells. Thus, if blocking the activity of any such proteins caused cancer cells to become resistant to thapsigargin-induced cell death, those proteins must be involved in the pathways that connect ER stress to apoptosis in the cancer cells.
The results revealed that thapsigargin-induced cancer cell death depended on two proteins called death receptor 5 and caspase-8. These proteins are both known to play important roles in apoptosis, with death receptor 5 acting upstream of caspase-8. Intriguingly, optimal caspase-8 activation and cell death induction by thapsigargin required the protein LC3B, which normally is associated with autophagy (a type of self-eating process that cells use to recycle their own components), but which in this case was involved in apoptosis induction via a non-autophagic function. The researchers further pinpointed how the various components of the unfolded protein response regulate cell death via influencing the expression levels of death receptor 5 and LC3B.
Interestingly, the researchers also found that one protein named IRE1 was required for thapsigargin-induced cell death in the prostate cancer cells, but not in the colon cancer cells. This demonstrates that the important players in regulating cell death and survival can often differ between different cancers, and that scientists need a good understanding of the factors driving tumor growth in all different cancers in order to treat patients effectively. The researchers’ results indicated an explanation for the cell type-dependent difference in that IRE1 acted via the JNK signaling pathway in a cytotoxic manner in the prostate cancer cells, whereas thapsigargin-induced cell death was independent of JNK in the colon cancer cells.
The findings provide new insights into the induction of cell death by ER stressors such as thapsigargin, and suggest that this compound could serve as a basis for new drugs designed for targeted killing of cancer cells.
The publication can be read in full on the Springer website.
A video accompanying the publication and explaining the major findings is available on the Vimeo website.