A recent publication by NCMM studied how the ER stressor drug thapsigargin and analogues for cancer-related therapies induce cell death in human cancer cells.
A recent publication by Lindner et al. entitled; “Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components” studied how the ER stressor drug thapsigargin and analogues for cancer-related therapies induce cell death in human cancer cells.
The collaborative work between researchers at NCMM, DANDRITE, the University of Copenhagen and Aarhus University provide important insights for the treatment of cancer by activating a process in cells known as the ‘unfolded protein response’.
Protein folding and upholding cell function
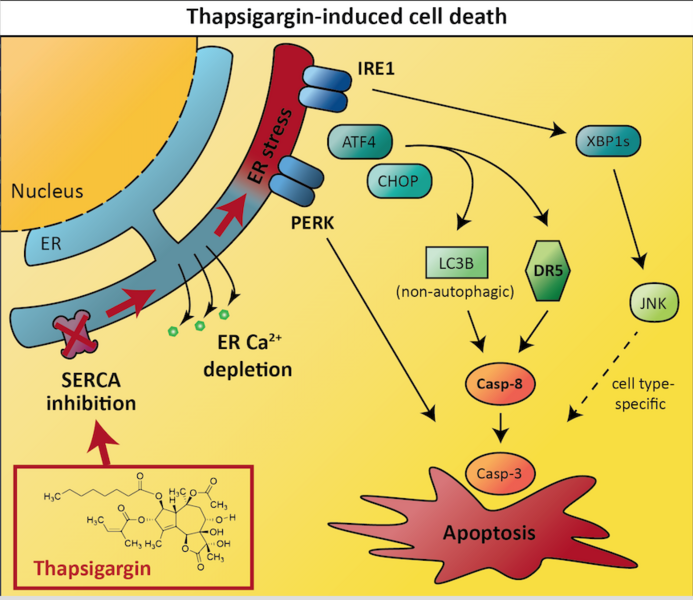
Each one of our cells contains thousands of protein molecules that together play a vital part in cell behaviour, function and survival. These protein molecules must be correctly folded in order to function optimally and maintain cell homeostasis as failure to control protein folding can lead to dysfunctional proteins and may be toxic to the cell. The synthesis of new proteins and the degradation of old proteins is an ongoing cycle in our cells. The endoplasmic reticulum (ER) is a network of membranous tubules within the cytoplasm and continuous to the nuclear membrane in our cells. The ER ensures that newly synthesized proteins in the cell are correctly folded. But, when the number of new protein molecules produced by the cell outdoes the ER’s capability to fold them, a process known as ER stress can occur. ER stress is related to the unfolded protein response (UPR) that is triggered in response to an accumulation of unfolded or misfolded proteins in the ER. This response is activated to help return the cell back to normal. Initially the first step is to re-establish balance in the cell by halting protein translation and the degradation and removal of misfolded protein is amplified. In addition, the production of chaperons that assist with conformational folding/unfolding and assembly/disassembly of proteins is also increased. If this step fails, the UPR aims towards apoptosis – a tightly controlled programmed cell death.
Activating ER stress and thereby UPR in human cancer cells
Inducing cell death via ER stress and the UPR is one strategy to target cancer, however the signalling mechanisms underlying UPR-mediated activation of cell death are incompletely understood. These molecular mechanisms need to be fully understood to design drugs that effectively trigger the UPR in cancer cells. The team of researchers provide new insights that improve our understanding of the UPR signalling mechanisms in cancer cells. Thapsigargin is a drug that can induce UPR-mediated cell death, but the detailed mechanisms remain unclear. Lindner et al. identified which cellular proteins that were essential for thapsigargin to initiate cancer cell death. The team systematically exposed human (LNCaP) prostate and (HCT116) colorectal cancer cell lines to thapsigargin or its analogues combined with the inactivation of a variety proteins in cancer cells, and were thereby able to identify two proteins (death receptor 5 and caspase-8) that are required for thapsigargin to effectively induce apoptosis in cancer cells. In addition, they found that LC3B, a protein normally associated with autophagy, was essential for optimal activation of caspase-8 and induction of apoptosis by thapsigargin. The expression of both death receptor 5 and LC3B was controlled by ATF4 and CHOP transcription factors of the unfolded protein response, whereas PERK, which normally acts upstream of ATF4 and CHOP, was required for thapsigargin-induced cell death, but not for the expression of death receptor 5 or LC3B. The team also succeeded in finding a protein named IRE1 that was specifically required for thapsigargin-induced apoptosis in the LNCaP prostate cell line and not in the HCT116 colorectal cell line. They found that IRE1 activated JNK signalling pathway via XBP1, and that JNK was required for thapsigargin-induced apoptosis in the prostate cancer cells, whereas thapsigargin-induced cell death was independent of JNK in the colon cancer cells. These novel results provide insights into both general and more cell-type specific signalling mechanism of apoptosis induction by ER stressors such as thapsigargin, and propose that the drug could serve as a basis for the development of targeted cancer therapy.
Find the full-text paper here.
Find a video abstract accompanying the publication on the Vimeo website.
Find the news story published by NCMM here.